Generating Contact Matrix
There are two common formats for contact maps, the Cooler format and Hic format. Both are compressed and sparsed formats to avoid large storage volumes; For a given \(n\) number of bins in the genome, the size of the matrix would be \(n^2\), in addition, typically more than one resolution (bin size) is being used.
In this section we will guide you on how to generate both matrices types, HiC and cool based on the .pairs file that you generated in the previous section and how to visualize them.
Generating HiC contact maps using Juicer tools
Additional Dependencies
Juicer Tools - Download the JAR file for juicertools and place it in the same directory as this repository and name it as
juicertools.jar. You can find the link to the most recent version of Juicer tools here e.g.:
wget https://s3.amazonaws.com/hicfiles.tc4ga.com/public/juicer/juicer_tools_1.22.01.jar
mv juicer_tools_1.22.01.jar ./HiChiP/juicertools.jar
Java - If not already installed, you can install Java as follows:
sudo apt install default-jre
From .pairs to .hic contact matrix
Juicer Tools is used to convert
.pairsfile into a HiC contact matrix.HiCis highly compressed binary representation of the contact matrixProvides rapid random access to any genomic region matrix
Stores contact matrix at 9 different resolutions (2.5M, 1M, 500K, 250K, 100K, 50K, 25K, 10K, and 5K)
Can be programmatically manipulated using straw python API
The .pairs file that you generated in the From fastq to final valid pairs bam file section can be used directly with Juicer tools to generate the HiC contact matrix:
Parameter |
Function |
|---|---|
-Xmx |
The flag Xmx specifies the maximum memory allocation pool for a Java virtual machine, from our experience 48000m works well when processing human data sets. If you are not sure how much memory your system has, run the command |
Djava.awt.headless=true |
Java is ran in a headless mode when the application does not interact with a user (if not specified, the default is Djava.awt.headless=false) |
pre |
The pre command allows users to create .hic files from their own data |
–threads |
Specifies the numbers of threads to be used (integer number) |
*.pairs or *.pairs.gz |
input file for generating the contact matrix |
*.genome |
genome file, listing the chromosomes and their sizes |
*.hic |
hic output file, containing the contact matrix |
Tip no.1
Please note that if you have an older vesrion of Juicer tools, generating contact map directly from .pairs file may not be supported. We recommend updating to a newer version. As we tested, the pre utility of the version 1.22.01 support the .pairs to HiC function.
Command:
java -Xmx<memory> -Djava.awt.headless=true -jar <path_to_juicer_tools.jar> pre --threads <no_of_threads> <mapped.pairs> <contact-map.hic> <ref.genome>
Example:
java -Xmx48000m -Djava.awt.headless=true -jar ./HiChiP/juicertools.jar pre --threads 16 mapped.pairs contact_map.hic hg38.genome
Tip no.2
Juicer tools offers additional functions that were not discussed here, including matrix normalization and generating matrix for only specified regions in the genome. To learn more about advanced options, please refer to the Juicer Tools documentation.
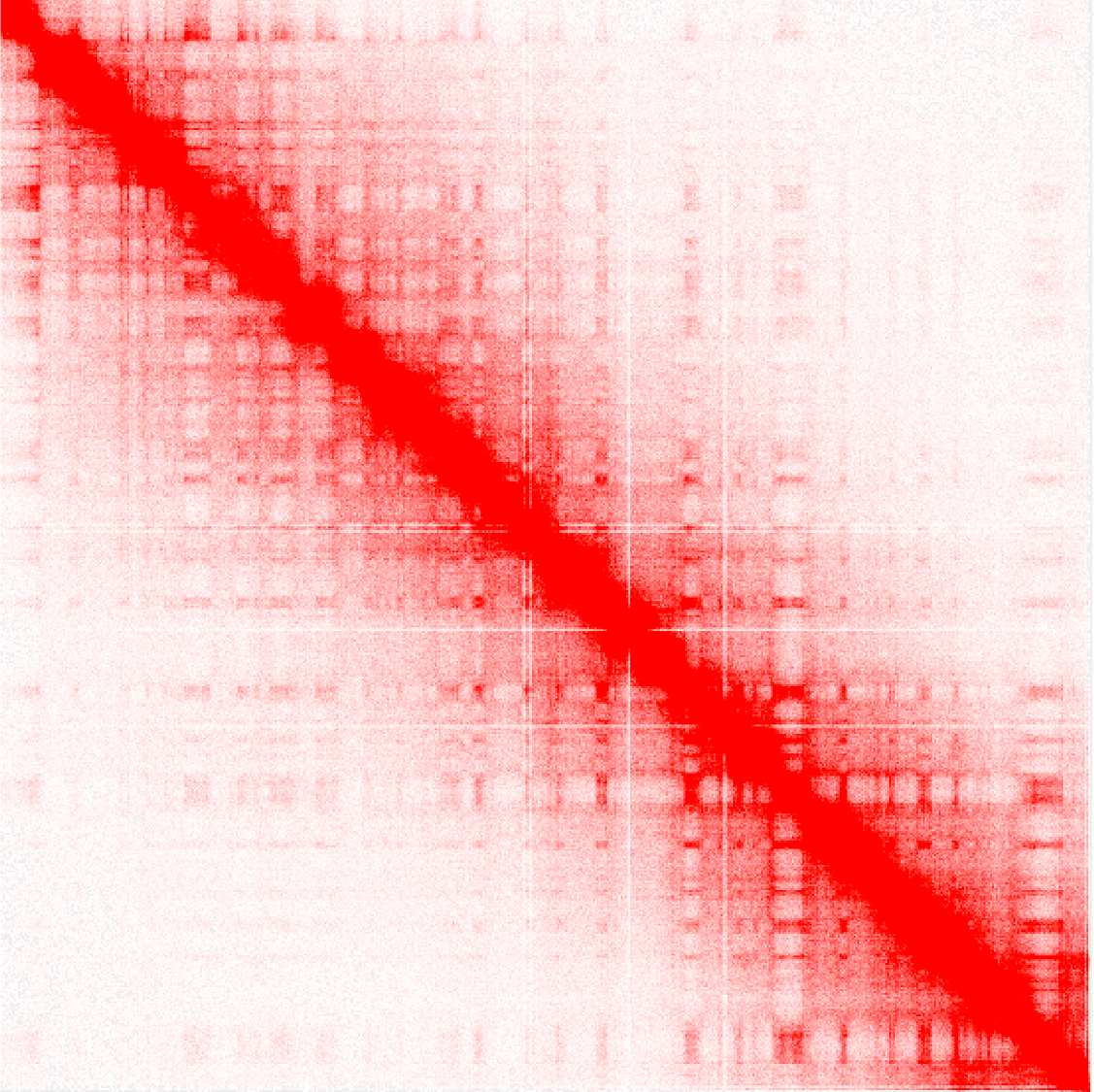
Visualizing .hic contact matrix
The visualization tool Juicebox can be used to visualize the contact matrix. You can either download a local version of the tool to your computer as a Java application or use a web version of Juicebox. Load your .hic file to visualize the contact map and zoom in to areas of interest.
Generating cooler contact maps
Additional Dependencies
Installing Cooler and its dependencies
For any issues with cooler installation or its dependencies, please refer to the cooler installation documentation
Installing Pairix
Pairix is a tool for indexing and querying on a block-compressed text file containing pairs of genomic coordinates. You can install it directly from its github repository as follows:
git clone https://github.com/4dn-dcic/pairix
cd pairix
make
Add the bin path, and utils path to PATH and exit the folder:
PATH=~/pairix/bin/:~/pairix/util:~/pairix/bin/pairix:$PATH
cd ..
Important!
make sure to modify the following example with the path to your pairix installation folder. If you are not sure what is the path you can check it with the command pwd when located in the pairix folder.
For any issues with pairix, please refer to the pairix documentation
From .pairs to cooler contact matrix
Cooler tools is used to convert indexed
.pairsfile into cool and mcool contact matricesCoolergenerates a sparse, compressed, and binary persistent representation of proximity ligation contact matrixStore matrix as HDF5 file object
Provides python API to manipulate contact matrix
Each cooler matrix is computed at a specific resolution
Multi-cool (mcool) files store a set of cooler files into a single HDF5 file object
Multi-cool files are helpful for visualization
Indexing the .pairs file
We will use the cload pairix utility of Cooler to generate contact maps. This utility requires the .pairs file to be indexed.
Pairix is used for indexing compressed .pairs files. The files should be compresses with bgzip (which should already be installed on your machine). If your .pairs file is not yet bgzip compressed, first compress it as follows:
Command:
bgzip <mapped.pairs>
Example:
bgzip mapped.pairs
Following this command mapped.pairs will be replaced with its compressed form mapped.pairs.gz
Note!
Compressing the .pairs file with gzip instead of bgzip will also result in a compressed file with the .gz suffix, but due to format differnces it will not be accepted as an input for pairix.
Next, index the file .pairs.gz file:
Command:
pairix <mapped.pairs.gz>
Example:
pairix mapped.pairs.gz
Genereting single resolution contact map files
As mentioned above, we will use the cload pairix utility of Cooler to generate contact maps:
cooler cload pairix usage:
Parameter |
Function |
|---|---|
<genome_fils>:<bin size> |
Specifies the reference .genome file, followed with``:`` and the desired bin size in bp |
-p |
Number of processes to split the work between (integer), default: 8 |
*.pairs.gz |
Path to |
*.cool |
Name of output file |
Command:
cooler cload pairix -p <cores> <ref.genome>:<bin_size_in_bp> <mapped.pairs.gz> <matrix.cool>
Example:
cooler cload pairix -p 16 hg38.genome:1000 mapped.pairs.gz matrix_1kb.cool
Genereting multi-resolutions files and visualizing the contact matrix
When you wish to visualize the contact matrix, it is highly recommended to generate a multi-resolution .mcool file to allow zooming in and out to inspect regions of interest. The cooler zoomify utility allows you to generate a multi-resolution cooler file by coarsening. The input to cooler zoomify is a single resolution .cool file, to allow zooming in into regoins of interest we suggest to generate a .cool file with a small bin size, e.g. 1kb. Multi-resolution files uses the suffix .mcool.
cooler zoomify usage:
Parameter |
Function |
|---|---|
–balance |
Apply balancing to each zoom level. Off by default |
-p |
Number of processes to use for batch processing chunks of pixels, default: 1 |
*.cool |
Name of contact matrix input file |
Command:*
cooler zoomify --balance -p <cores> <matrix.cool>
Example:
cooler zoomify --balance -p 16 matrix_1kb.cool
The example above will result in a new file named matrix_1kb.mcool (no need to specify output name)
Tip
Cooler offers additional functions that were not discussed here, including generating a cooler from a pre-binned matrix, matrix normalization and more. To learn more about advanced options, please refer to the cooler documentation
HiGlass is an interactive tool for visualizing .mcool files. To learn more about how to set up and use HiGlass follow the HiGlass tutorial