Welcome to HiChIP documentation
Overview
The Dovetail™ HiChIP MNase Kit combines the benefits of ChIP-seq with Hi-C, a proximity ligation method that captures long-range interactions using standard Illumina paired-end sequencing, enabling researchers to query protein-directed chromatin conformation mediated by specific proteins of interest.
Key benefits of HiChIP:
Capture ChIP-seq and Hi-C data together in a single library
Map chromatin interactions at nucleosome level resolution
The unique combination of the Dovetail™ Micro-C Proximity Ligation Assay with the Dovetail HiChIP approach enables the use of micrococcal nuclease (MNase) to fragment chromatin uniformly and without sequence bias prior to proximity ligation, eliminating the need for finicky sonication procedures and offering the maximal resolution (down to mono-nucleosome size) of chromatin interactions.
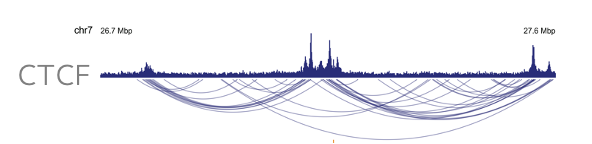
Enrichment of protein-directed chromatin features enables high-resolution contact map generation with less read depth. Compared to a high resolution restriction enzyme-based Hi-C, Dovetail HiChIP data enables visualization of higher-order chromatin features, such as loops and chromatin interactions, at a fraction of the read depth leading to significant sequencing costs savings.
This guide will take you step by step on how to QC your HiChIP library, how to interparate the QC results and how to call and plot significant interactions. If you don’t yet have a sequenced HiChIP library and you want to get familiar with the data, you can download HiChIP sequences libraries from our publicaly available data sets.
The QC process starts with aligning the reads to a reference genome then retaining high quality mapped reads. From there the mapped data will be used to generating a pairs file with pairtools, which categorizes pairs by read type and insert distance, this step both flags and removes PCR duplicates. Once pairs are categorized, counts of each class are summed and reported.
If this is your first time following this tutorial, please check the Before you begin page first.
Contents: