HiChIP Loop Calling
Introduction
This workflow is a simple guide to identify loops in HiChIP data. Before you get started please read this short introduction which will help you better understand what loops are in the context of HiChIP assays and why we’re going to focus on FitHiChIP tool as the tool to use. I would like preface this work by saying there is no “one correct way” to analyze HiChIP data. This is just an example workflow that will enable you identify significant interactions in HiChIP data. The biological implications of those interactions should be interpreted through the lenses of the protein target or biological question you’re asking!
What are chromatin Loops in the context of HiChIP?
HiChIP loops are - significant interactions between a protein-anchor and the surrounding genome.
The biological interpretations of the interactions is based on the protein of interest for example:
CTCF – Identifying actual chromatin loops. Meaning, CTCF-cohesin mediated looping.
H3K27ac or H3K4me3 – means identifying regions that interact with the enhancer or active promotor marker respectively. These interactions do not necessarily reflect the canonical loop formation but could reflect short range folding.
As these interactions can reflect more than just canonical “loops”, they will simply be referred to as significant interactions for the rest of the documentation.
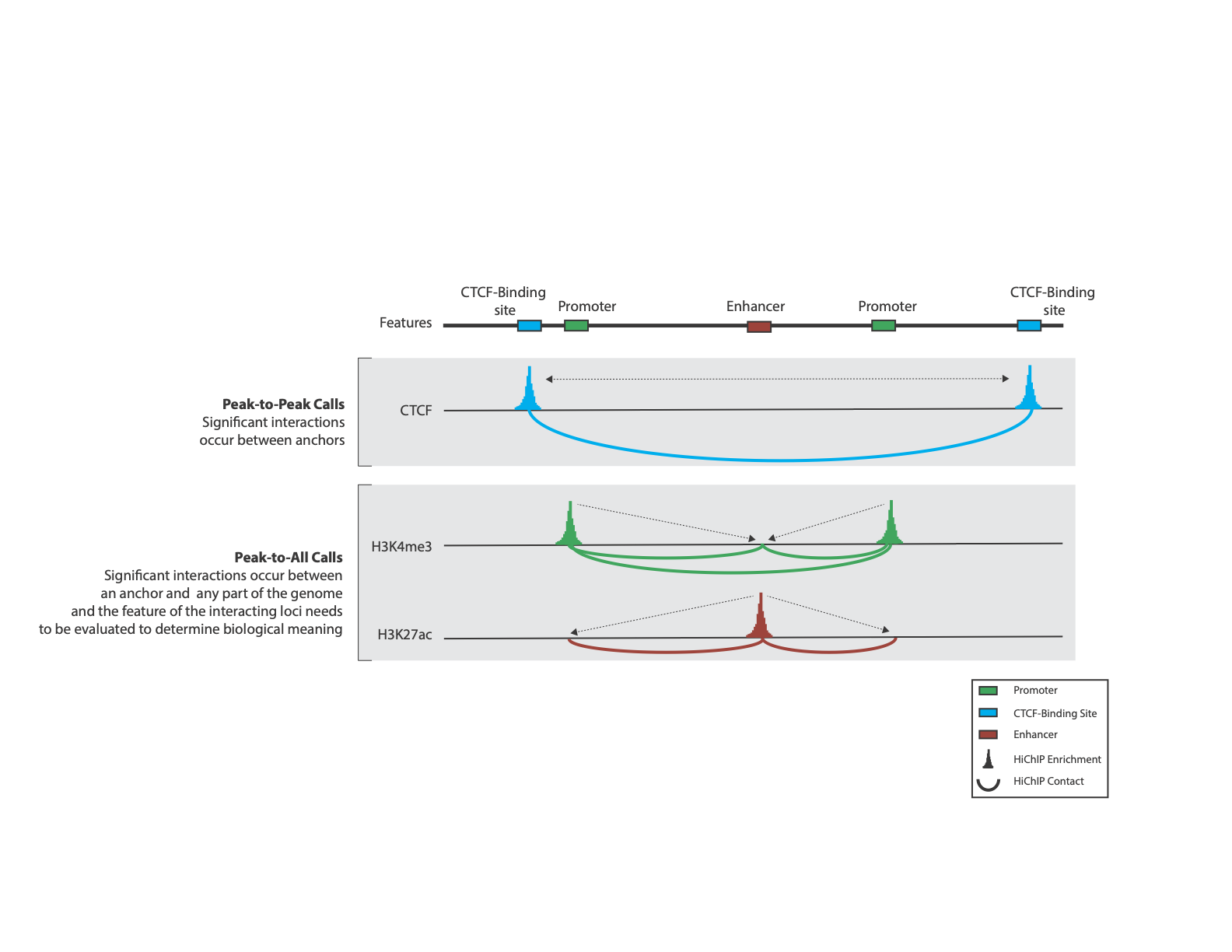
Types of significant HiChIP interactions
Peak-to-Peak - ChIP-seq anchors will only interact with other anchors like CTCF chromatin loops
Peak-to-All - ChIP-seq anchors will interact with any of the surrounding genome weither or those regions correspond with an anchor site, like H3K27ac, H3K4me3, or PolII
All-to-All - This is for a non-targeted Hi-C data contact matrix and will not be used here.
HiChIP interactions type:
Resolution
Resolution can play an odd roll in HiChIP significant interaction detection. Typically, only 5-10 reads-pairs per interaction are required to statistically identify an interaction with HiChIP as opposed to the 100-1000 read-pairs required for non-targeted Hi-C assays. However, many loop callers work on contact matrices which require binning at a specified resolution(s) to build. Typically, 1 kb, 5 kb, or 10 kb for HiChIP data. As such, the fewer reads you have, the larger the bin size must be to have enough read support to run statistics on.
Additionally, the biological nature of the protein target can impact the resolution of that you are interested in looking at. Proteins with a larger footprint (PolII) might require larger bin size, where smaller footprints (CTCF) might need a smaller bin. What becomes problematic, is when the bin size is so large that many anchors are captured in a single bin. This is important to keep in mind.
With all that being said, most HiChIP/ChIA-PET analyses are conducted between 2.5 - 5 kb. Our best recommendation is to try calling significant interactions at different resolutions. Generate lists of significant interactions at multiple resolutions and filter to keep only unique entries.
Tool landscape
There are many tools available to identify significant interactions. Below is a table that outlines just a subset of tools, where to get them, requirement to specify a resolution, ability to select the type of interaction, and input file structure of the HiChIP data. They all have their own pro’s and con’s, but there is no clearly established way to analyze HiChIP data, and it largely depends on your biological questions. So always keep that in mind and make sure the tool you’re using makes sense with the biological question you are asking!
Tool |
Repo |
Input format |
Resolution Option |
Considers type of interaction |
|---|---|---|---|---|
FitHiChIP |
HiC-Pro ValidPairs |
Yes |
Yes |
|
Cloops |
Bedpe |
No |
No |
|
HiChIPPER |
HiC-Pro ValidPairs |
Yes |
No |
|
HiCCUPs |
.hic |
Yes |
No |
Why FitHiChIP?
We have chosen FitHiChIP for this workflow for a few reasons:
The install is very easy, and it can manage all the dependencies through Docker or Singularity (if you don’t have sudo privileges)
It is very flexible in term of input,
.pairs, or interaction tabel in.bedpeformat.Has the ability to select bias type
Can specify the type of interaction to assess
Output is easily integrate-able to other workflows
Below is an annotated configuration file with some of the key parameters to consider

Input files
This workflow assumes you have completed the Step-by-step guide to Process HiChIP data. The two key files required are:
Filtered Pairs file - output from From fastq to final valid pairs workflow.
Bed file of ChIP-seq anchors for your protein of interest, e.g. as you used in the QC step. We included in the datasets section links to some useful ChIP-seq bed files from the Encode project.
Testing!
If you are looking for a dataset to practice this walkthrough, I reccomend the GM12878 CTCF (deep sequencing) from our publicaly available datasets
Tools
Workflow Overview
Run FitHiChIP through docker - FitHiChIP is a single executable that:
Builds a table of interactions (bedpe-like version of a contact matrix)
Corrects for biases (coverage or ICE)
Filters data for the type of interactions (Peak-to-Peak, Peak-to-All, or All-to-All)
Builds a contact frequency to insert distance model from the filtered interactions.
Assigns P-values and Q-values (false discovery rate - FDR) to interactions.
Will merge near-by interaction that pass a Q-value threshold.
Report a bedpe-like file of total and merged interactions filtered by a Q value.
Workflow
Convert filtered pairs file to Hi-C Pro valid pairs format
Command:
grep -v '#' <*.pairs>| awk -F"\t" '{print $1"\t"$2"\t"$3"\t"$6"\t"$4"\t"$5"\t"$7}' | gzip -c > <output.pairs.gz>
Example:
grep -v '#' mapped.pairs| awk -F"\t" '{print $1"\t"$2"\t"$3"\t"$6"\t"$4"\t"$5"\t"$7}' | gzip -c > hicpro_mapped.pairs.gz
Modify the
configuration fileto desired specifications:We’ll be using coverage bias because these data are MNase based, not RE-based
If using CTCT use Peak-to-Peak as outlined earlier, CTCF data is a peak to peak interaction, other protein like H3K27ac and H3K4me3 you’re going to want to use Peak-to-All.
Adjusting the configuration file . Entries that need to be adjusted are highlighted:
#====================================
# Sample configuration file for running FitHiChIP
#====================================
#*****************************
# important parameters
#*****************************
# File containing the valid pairs from HiCPro pipeline
# Can be either a text file, or a gzipped text file
ValidPairs=/path_to_hicpro_pairs/prefix.hicpro.valid.pairs.gz
# File containing the bin intervals (according to a specified bin size)
# which is an output of HiC-pro pipeline
# If not provided, this is computed from the parameter 1
Interval=
# File storing the contact matrix (output of HiC-pro pipeline)
# should be accompanied with the parameter 2
# if not specified, computed from the parameter 1
Matrix=
# Pre-computed locus pair file
# of the format:
# chr1 start1 end1 chr2 start2 end2 contactcounts
Bed=
# File containing reference ChIP-seq / HiChIP peaks (in .bed format)
# mandatory parameter
PeakFile=/path_to_ChIP_peaks/peaks.bed
# Output base directory under which all results will be stored
OutDir=/path_to_output/fithichip_test_1kb
#Interaction type - 1: peak to peak 2: peak to non peak 3: peak to all (default) 4: all to all 5: everything from 1 to 4.
IntType=1
# Size of the bins [default = 5000], in bases, for detecting the interactions.
BINSIZE=2500
# Lower distance threshold of interaction between two segments
# (default = 20000 or 20 Kb)
LowDistThr=20000
# Upper distance threshold of interaction between two segments
# (default = 2000000 or 2 Mb)
UppDistThr=2000000
# Applicable only for peak to all output interactions - values: 0 / 1
# if 1, uses only peak to peak loops for background modeling - corresponds to FitHiChIP(S)
# if 0, uses both peak to peak and peak to nonpeak loops for background modeling - corresponds to FitHiChIP(L)
UseP2PBackgrnd=1
# parameter signifying the type of bias vector - values: 1 / 2
# 1: coverage bias regression 2: ICE bias regression
BiasType=1
# following parameter, if 1, means that merge filtering (corresponding to either FitHiChIP(L+M) or FitHiChIP(S+M))
# depending on the background model, would be employed. Otherwise (if 0), no merge filtering is employed. Default: 1
MergeInt=1
# FDR (q-value) threshold for loop significance
QVALUE=0.01
# File containing chromomosome size values corresponding to the reference genome.
ChrSizeFile=/path_to_genome_file/hg38.genome
# prefix string of all the output files (Default = 'FitHiChIP').
PREFIX=prefix.2.5kb
# Binary variable 1/0: if 1, overwrites any existing output file. otherwise (0), does not overwrite any output file.
OverWrite=1
Run FitHiChIP through docker
Command:
FitHiChIP_Docker.sh -C config.txt
Inspect the report

Output
FitHiChIP merged interactions output
What if?
I don’t have a bed file of ChIP-seq anchors or I can’t find a representative bed file for my antibody or sample type?
Follow our guide to calling 1-Demensional peaks with HiChIP data using MACS2
I want to use a different tool to identify significant interactions.
That is great! This is just one way please refer to tool you’d like to use for documentation. This is just one example of how to find significant interactions in HiChIP data. The key things to consider are the input formats of the data the tool requests.
I need to do differential analyses.
The output of this workflow is nice because the output is a bed file and if you have two samples one could just do a
bedtools intersectto classify interactions as shared or unique to each sample.
What next?
Visualization
Continue with plotting HiChIP interactions in R
Import to the Wash-U epigenome browser (more information in this link)